Medical Devices 101 // Pacemakers

DATE
November 03, 2020
AUTHOR
Alex | Product Manager
What is a pacemaker?
A pacemaker is a device that’s placed under the patient’s skin in their chest to help control their heartbeat if they have an irregular heartbeat (called “arrhythmia”), particularly a slow one.
According to Grand View Research, the global pacemaker market size was valued at USD 4.5 billion in 2019 and is expected to grow at a compound annual growth rate (CAGR) of 2.9% in the next seven years. There are many factors driving the market, such as an increasing number of incidences of Cardiovascular Diseases (CVDs), technological innovation and favorable government policies. The main reason for market growth, however, is the massive burden of CVDs. On the one hand, coronary heart disorder is one of the main causes of death due to CVDs globally. On the other hand, the financial burden is expected to be more than USD 1,044.0 billion by 2030, which highlights the importance of and necessity of these medical devices.
The pacemaker industry is – like most others – strongly affected by the COVID-19 pandemic. The novel disease also has a severe impact on patients suffering from CVDs. According to the Centers for Disease Control and Prevention, having a cardiovascular disease increases the risk of severe illness from COVID-19. Consequently, treatment of these diseases is becoming even more important.
As relatively high-risk class devices, pacemakers are typically classified as III. In this blog post, we will discuss different aspects of pacemakers and what manufacturers need to be aware of under EU MDR law.
How are pacemakers inserted?
Inserting a traditional pacemaker does not require open heart surgery and most patients are released from hospital within 24 hours. During the surgery, a small incision is made in the upper part of the patient’s chest. A thin insulated wire (or “lead”), is then guided through the vein into the patient’s heart. Following this, the surgeon connects the lead to the pacemaker, which is inserted beneath the skin in the upper chest. Finally, the device is programmed and tested to ensure it’s working properly. As a final step, the incision is closed.
What types of pacemaker are there?
There are three main types of pacemakers, which are selected based on the patient’s symptoms and specific heart condition.
- Single chamber pacemaker. This type has one lead, which connects to one chamber of the heart. In most cases, the pulse generator is connected to the right ventricle in the lower heart chamber.
- Dual chamber pacemaker. This type carries electrical impulses to the right ventricle and the right atrium of the patient’s heart to regulate the pace of contractions between the two chambers.
- Biventricular pacemaker. This pacemaker is also known as cardiac resynchronization therapy device. With three leads, it is connected to the right atrium and both ventricles. Biventricular pacemakers are used to treat people with arrhythmias caused by advanced heart failure.
What’s the new process for certifying and monitoring pacemakers under EU MDR?
Pacemakers are a class III medical device, which, according to the Johner Institut, account for about 2% of the medical device market share. When it comes to the conformity assessment and Post-Market Surveillance (PMS), pacemakers are subject to several important legal requirements. In the next paragraphs, we will examine how the EU MDR will change this process, which will replace the MDD (Medical Device Directive) in May 2021.
Conformity Assessment
The conformity assessment procedure proves that the general safety and performance requirements of the medical device are fulfilled and is primarily the responsibility of the medical device manufacturer. However, it is subject to the regulatory requirements established in the location where the device is sold, i.e. the EU MDR in Europe.
The conformity assessment procedure is described in Article 52 of the EU MDR. The type of assessment depends on the device’s risk class and specific features of certain devices.
According to the MDR, almost all medical device classes (except for class I), require a Notified Body. For pacemakers as a class III device, manufacturers either need a full Quality Management System (QMS) compliant with ISO 13485, which is specified in Annex IX, or they have to follow Annex XI.
For medical devices of classes IIb and III, an additional examination is necessary, which is detailed in Annex X of the EU MDR. However, this is only needed if Annex IX is not followed i.e. the manufacturer does not have a full QMS system). This “type-examination” is the procedure whereby the Notified Body verifies that the pacemaker complies with the relevant provisions.
Post-Market Surveillance
Post-Market Surveillance (PMS), is the practice of monitoring the safety and performance of a device once it has been taken to market. This is important, as certain risks or flaws of a pacemaker might only become apparent at a later stage. It is therefore crucial to continuously collect post-market data on a device’s performance, as this will ensure a high standard of product quality.
In contrast to the MDD, which only briefly mentions the need for PMS, the MDR provides detailed requirements for a PMS plan in Article 82. For class III medical devices such as pacemakers, the manufacturer must establish and maintain a PMS system to actively and systematically gather, record and analyse data on the quality, performance and safety of the product throughout its entire lifecycle, This enables manufacturers to quickly initiate any necessary measures such as product callbacks in case of any (serious) adverse events.
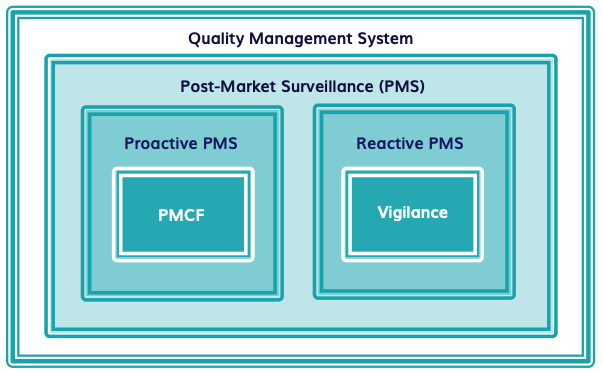
When it comes to PMS, there are two types: proactive and reactive PMS. Reactive PMS, also called ‘Vigilance’, describes the response to a specific incident, such as a (serious) adverse event. For pacemakers, these could include infection at the surgical site, sensitivity to the device material or failure to deliver therapy. In contrast, proactive PMS tries to anticipate and prevent incidents by collecting data about the device’s performance. This can include surveys among end users or customers, post-CE mark clinical studies or user groups.
Navigating Post-Market Surveillance in the EU MDR Era
Clinical Evaluation Report
The Clinical Evaluation Report (CER) documents the findings of a clinical evaluation of the pacemaker and is an essential step to obtaining the CE certification. The report consists of analysed clinical data that was either collected from a clinical investigation of the own device or results from previous literature reviews on substantially equivalent devices.
The EU MDR introduced new guidelines for CER reports, which can be found in Article 61 and Annex XIV. In the MDR, the scope and emphasis of the CER is more important compared to the MDD.
The Clinical Evaluation Report must be updated throughout a device’s lifecycle. As pacemakers are a class III and therefore high-risk device, the report must be updated at least annually.
Periodic Update Safety Report
Under MDR law, manufacturers must also regularly submit a Periodic Update Safety Report (PSUR). This report provides authorities with a summary of the results and conclusions from the gathered PMS data. Manufacturers of class III devices, such as pacemakers, must update the PSUR when needed and at least annually, according to Article 86 of the MDR.
A PSUR should contain the following:
- Meta information including references
- Plan implementation
- Product data (including sales, indications, patients)
- Overview of data (including adverse events)
- Results of analysis (including trends)
- Overview of actions taken (including CAPA, vigilance, FSCA)
- Conclusions from the risk-benefit ratio
- Final assessment
Source: Johner Institut.
Post-Market Clinical Follow-up
Finally, Post-Market Clinical Follow-up (PMCF) is part of PMS and is also a mandatory process according to the EU MDR. As defined in Annex XIV Part B, PMCF is “a continuous process that updates the clinical evaluation […] and shall be addressed in the manufacturer’s post-market surveillance plan.” In other words, PMCF’s aim is to continuously confirm the safety and performance of the pacemaker. The clinical data collected on the pacemaker should demonstrate that its risks remain acceptable in the context of its clinical benefit.
PMCF activities can range from analysing data from publicly available information in existing device registrations to performing a new clinical investigation. These investigations can be done in the form of digital surveys, as we will discuss in the next section.
Data collection – time to take it digital
As described in the previous sections, PMS activities require continuous and systematic data collection throughout the entire lifetime of the pacemaker. Therefore, a significant amount of clinical data is required, which can pose a challenge for many manufacturers. In addition, the EU MDR will bring about some significant changes, including more rigorous controls on clinical reports and higher expectations regarding the collection of clinical data for medical devices and their CE marking,
In a recent survey about the true cost of the EU MDR, we asked participants how they are currently managing their PMS activities including their PMCF studies. The results showed that 69% use Excel sheets, 47% use paper and only 11% have digialized their data collection with an Electronic Data Capture solution
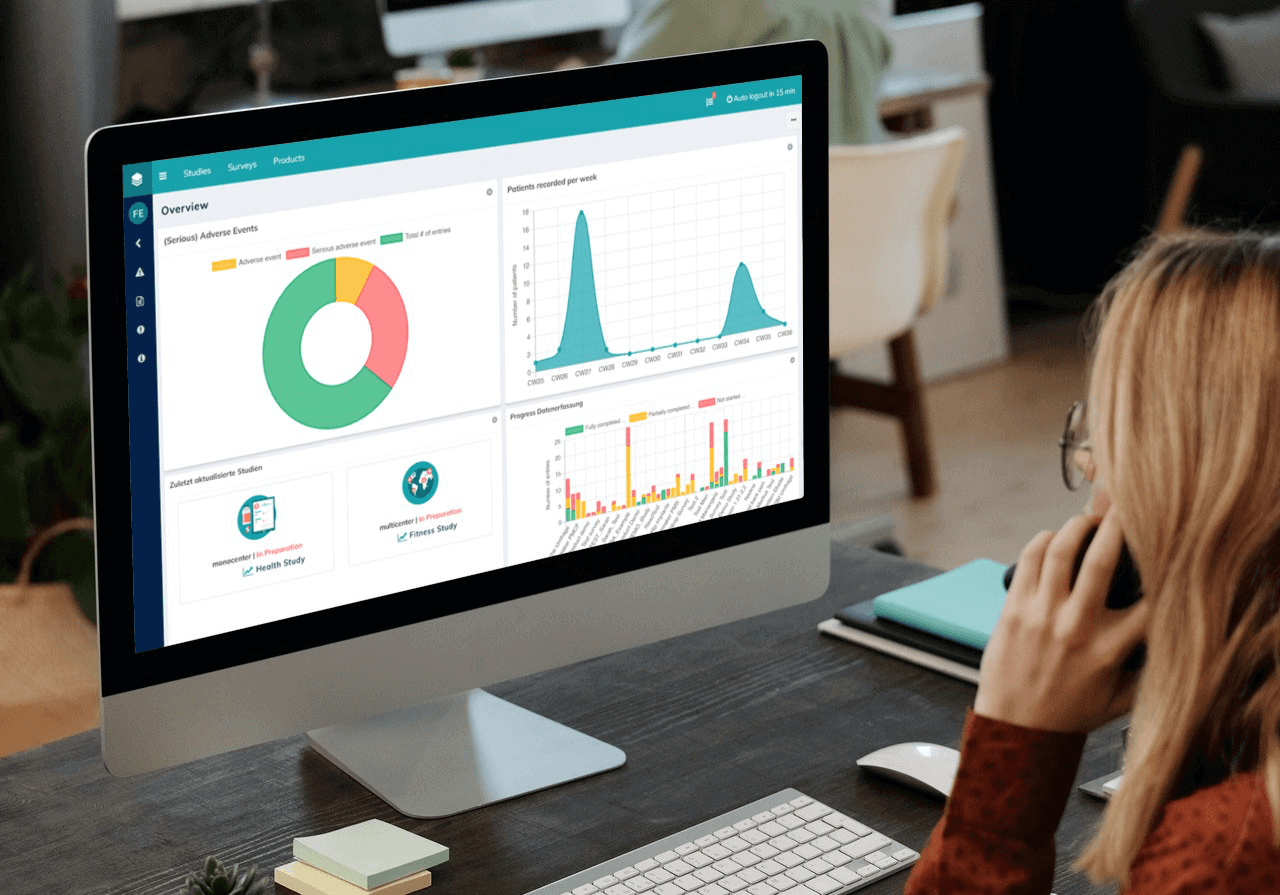
Using Electronic Data Capture (EDC) systems i.e. capturing and managing data on a digital platform, is one of the most efficient ways to conduct your PMCF activities in compliance with the EU MDR. Considering the profound regulatory changes the EU MDR entails, we have good reason to believe that paper-based data collection methods will be impossible to sustain, as manufacturers relying on these methods will likely struggle to meet the data collection requirements in the near future.
EDC solutions allow the manufacturer to collect data in real-time and display them in interactive dashboards, which offeran overview of all current measures and metrics. Such a solution also enables smooth collaboration between all involved parties – such as doctors, patients or expert user groups for PMS activities.
Discover the power of EDC solutions for yourself
Want to learn more about how you can digitalize your PMS activities? Get your free trial here:
Survey example for pacemaker
If you’d like to see a survey example for pacemaker in action, take a look at this PMCF survey which was created with Climedo’s software.
Alternatively, scan this QR code with your smartphone to access the survey:


Alex | Product Manager
Climedo
Enthusiastic about the innovation and possibilities of the health tech industry, given her background in biomedicine. Passionate about traveling, sports and photography.
You may also like these articles