Getting your Post-Market Surveillance (PMS) up to Speed with the EU MDR

DATE
November 09, 2019
AUTHOR
Catherine | Associate Director Marketing
This article first appeared on Medium.
In April 2017, the European Commission adopted a new Regulation (EU MDR 2017/745) on medical devices, which will progressively replace current directives, such as the MDD (Medical Devices Directive). The transitional period for implementing the requirements ends in May 2021. This regulation has major implications on how PMS (Post-Market Surveillance) and PMCF (Post-Market Clinical Follow-up) will be carried out by medical device manufacturers in the future. We recently took a deep dive into what changes will be required from manufacturers and offer a 7-point action plan for getting processes and activities up to speed with the EU MDR. The findings are presented in our Whitepaper “Navigating Post-Market Surveillance in the EU MDR Era” which you can download for free here.
And for those interested, the full 117-page regulation can be found here in 24 languages.
Why the new regulation?
Under the EU MDR, manufacturers across all member states of the European Union (EU) — and any organization selling devices within the EU — must make significant changes in their product development, data reporting and quality assurance.
One of the long-term objectives of the MDR is to better observe and track how devices are actually being used by patients and medical staff on the market. It aims to establish systematic evidence of whether a device is fulfilling its intended purpose, (e.g. is it being underused or overused, are the instructions clear?), and whether it is still safe for the patient. Competing devices must also be tracked and compared, which will help to justify whether a particular device is still valuable enough on the market or needs to be discontinued. In terms of risk management, the benefits of using a device must always outweigh the potential risks it carries. As such, the MDR aims to create and establish a modernized and robust framework to ensure that public health and patient safety. Therefore, key elements of the existing approach will be enforced, including vigilance and market surveillance, while ensuring transparency and traceability.
What does this mean for medical device manufacturers?
For manufacturers, adapting to the new regulation will entail higher costs and longer timelines for their product development. There will also be new clinical monitoring and evidence generation processes to recertify existing products.
Should manufacturers fail to carry out the required Post-Market Surveillance (PMS) activities, they risk losing their ISO 13485 certification, having their devices taken off the market, being fined or even prosecuted. In fact, German and Swiss authorities predict that 50% of all medical devices will be discontinued or fail to meet the MDR requirements.
As the deadline for implementing the new requirements approaches, many manufacturers feel overwhelmed by the plethora of demands awaiting them, particularly with regard to PMS. The United States has even reached out to the EU asking for an MDR deadline extension — so far, there has been no official response by the EU. The EUDAMED database launch, however, has been postponed by two years until May 2022, coinciding with the deadline for implementing IVDR requirements.
Understanding PMS and PMCF
Medical device manufacturers are obliged to set up a systematic PMS approach as part of their Quality Management System (QMS). According to Chapter VII of the MDR, the surveillance system must be planned, set up, documented and continuously updated. Market surveillance data collection will serve to confirm and monitor the benefit-risk profile of the product and must be submitted to the Notified Body (NB) for inspection as part of a regularly updated report (Periodic Safety Update Report — PSUR). After May 2021, these reports will be made available to patients, users and regulatory authorities via the European Database for Medical Devices (EUDAMED).
Post-Market Surveillance (PMS) is a collection of processes and activities which are used to monitor the performance of a medical device in the market. The purpose of these activities is to gather information about the use of the device in order to identify potential device design and/or usage problems, as well as to accurately characterize the real-world device behavior and clinical outcomes. PMS needs to be set up as a system that provides continuous and proactive feedback about a device on the market in order to maintain a high standard of product quality.
Post-Market Clinical Follow Up (PMCF), on the other hand, is a subset of PMS. It is a continuous process in the scope of a clinical study that updates the clinical evaluation, i.e. the assessment and analysis of clinical data that pertains to a medical device in order to verify its clinical safety and performance when applied as intended by the manufacturer. When conducting PMCF, manufacturers must proactively collect and evaluate clinical data with the goal of confirming the safety and performance throughout the expected lifetime of their device, ensuring the acceptability of identified potential risks, and detecting emerging risks based on factual evidence.
What should your PMS and PMCF plans include?

We detail specific steps that manufacturers must take in terms of their PMS and PMCF activities in our new Whitepaper. We also offer an outlook and some of the advantages the regulation will bring to the industry in the long term. Want to find out more?
Understanding the Post-Market Surveillance Process
This infographic details the steps you need to be aware of when planning your Post-Market Surveillance Process.
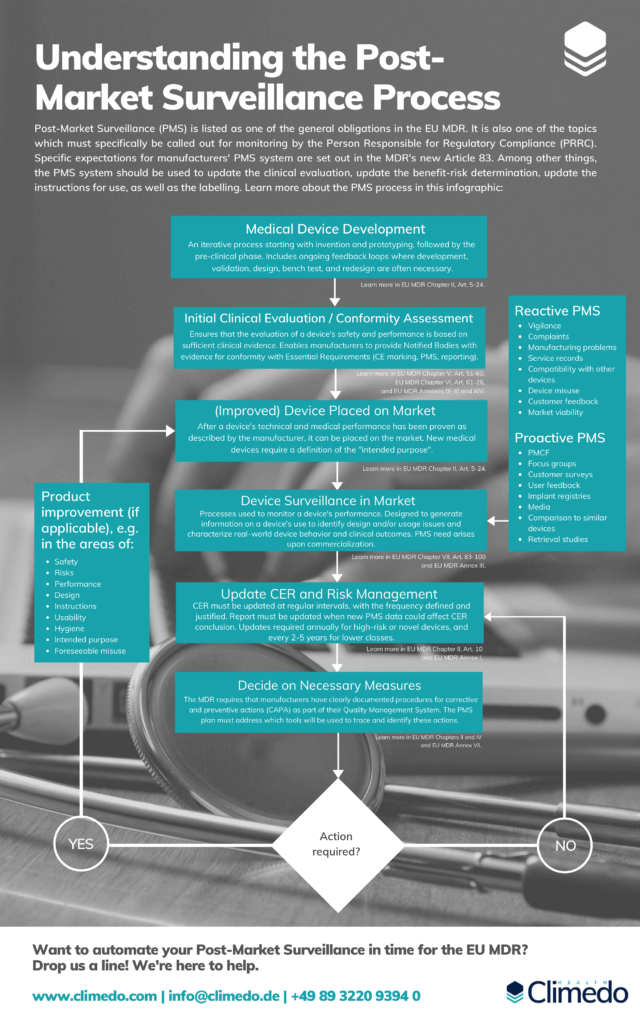
And if you’d like a free demo of our software that helps you automate PMS and PMCF activities in a standardized way, get in touch!
Additional downloads to get you ready for the EU MDR

Catherine | Associate Director Marketing
Climedo
Passionate about creating innovative event and content marketing formats to make the benefits of digital health solutions more tangible. Enjoys fine art, photography, foreign languages and all things digital.
You may also like these articles



