Feeling Unlucky on Friday 13th? Don’t Make these 6 PMCF Mistakes!

DATE
November 13, 2020
AUTHOR
Dragan | Co-Founder & CTO
Friday 13th doesn’t have to be a bad thing! If you’re worried about something going wrong with your clinical studies and surveys today, take a look at these six PMCF mistakes to avoid when conducting your post-market surveillance activities under EU MDR law and how to get them right.
Introduction
According to the new European Medical Device Regulation (EU MDR), Post-Market Surveillance (PMS) activities will become crucial for identifying and investigating potential residual risks of medical devices after they have been taken to market. As a result, these risks should be tracked and assessed through a systematic Post-Market Clinical Followup (PMCF) plan that is integrated within medical device manufacturers’ Quality Management System (QMS).
PMCF is required for all classes of medical devices and implants. As the MDR deadline in May 2021 approaches, manufacturers with devices which had previously obtained compliance based on equivalence with another device on the market, will now require proactive PMCF studies conducted to reassess their residual risks in order to be compliant.
While PMCF will increase costs of PMS in the short term, it can also be seen as an opportunity for alignment of processes across the operational portfolio in the long term. Thus, making the right initial choices is crucial, as a proactive PMCF system can be leveraged to not only gather data for compliance, but also to gain more comprehensive market insights and product marketing claims.
6 PMCF Pitfalls to avoid in the EU MDR Era
- Download our free Playbook on common PMCF mistakes
- Prepare yourself for potential consequences
- Learn how to get PMCF right

#1 Misinterpreting the Terms and Requirements
The Mistake
PMCF plans encompass a broad range of activities, one of which may be conducting a PMCF study from scratch. Manufacturers often do not understand the semantic differences between PMCF, PMCF plan and PMCF studies, and use them interchangeably. This can lead to missing or incorrect data and costly delays in (re-)certification.
The Consequences
PMCF under the MDR is a broad term that encompasses the collection of all types of clinical information such as vigilance, complaints, technical information, and publicly available information. However, the MEDDEV 2.7/1 rev 4, which is the primary guidance document for PMCF, places a strong focus on conducting PMCF studies and collecting data. It is therefore crucial to understand when PMCF studies are to be undertaken and when post-market data will suffice to avoid costly mistakes and wasting resources.
How to Get It Right
How does a manufacturer determine when market data is not sufficient and a PMCF study is required for fulfilling compliance? To help answer this, MEDDEV 2.7/1 rev 2 determines a set of limitations in clinical data in the pre-market phase. According to this document, limitations would include a limited number or relative homogeneity of subjects; a narrow diversity in study population; or an imbalance between use under controlled variables versus use under conditions of general medical practice. In these cases, PMCF is needed.
#2 Choosing the Wrong Study Design
The Mistake
As with any high-quality clinical investigation, it is essential for a PMCF study to have the right study design to adequately collect the required data. A common challenge of investigator-sponsored studies is that the sponsor or manufacturer may not be in control of how the study is conducted, including the correct use of the medical device within the study setting.
The Consequences
With the MDR deadline around the corner, many PMCF studies are being undertaken to fulfill compliance requirements, especially for devices of Class IIb or higher. It is imperative that the data produced by these studies can be used for evidence of compliance, otherwise manufacturers run the risk of incurring fines, having their devices taken off the market, or being refused recertification. This is particularly the case if manufacturers until now have only been using equivalence data for certification under the Medical Device Directive (MDD).
How to Get It Right
Manufacturers should ensure that their PMCF studies are conducted in strict compliance with the international clinical investigation standard, ISO 14155. The design of PMCF studies should clearly address the objective of the study, and the study hypothesis and endpoints should be scientifically sound, thus allowing for valid conclusions. At minimum, the PMCF study must define a rationale and justification of chosen study, study population (corresponding to the CE-mark scope), objectives of study, related study endpoints and statistical considerations, inclusion/exclusion criteria, subject number and follow up period of patients, and justification for selection of sites and investigators
#3 Selecting the Wrong Study Subjects
The Mistake
PMCF studies should always target the most important product user. For example, a PMCF study for a balloon catheter should rely on data submitted by surgeons, since these practitioners are generally the sole users of the devices. Conversely, when it comes to wearable devices, patients are the best subjects to submit PMCF data. PMCF studies can, however, be based on the wrong study population, often because a manufacturer is inexperienced in this field or selects of subjects based on ease-of-access rather than familiarity with a product.
The Consequences
Choosing an incorrect or irrelevant study population for a PMCF study can be time consuming and costly. Such studies often develop the burden of missing data and many users are lost in follow-ups, which further reduces the cost-effectiveness of PMCF activities.
How to Get It Right
Manufacturers should take steps to ensure that their chosen study population is appropriate for submitting valuable PMCF data. When PMCF study subjects are selected strategically and appropriately, the challenges of missing data and attrition are minimized, which, in turn, improves cost-effectiveness of PMCF activities. Employing a specialist team that includes statisticians and that is experienced in conducting such studies can be a reliable strategy.
#4 Misjudging Patient Recruitment Timelines
The Mistake
Manufacturers often fail to realize that, much like clinical investigations, PMCF studies can also suffer from challenges in patient enrolments, especially when it comes to accessing the appropriate patient populations and timelines.
The Consequences
Patient enrolment timelines are often underestimated by manufacturers, especially in the case of higher-class devices. Screening and recruiting patients who meet the carefully chosen inclusion or exclusion criteria for a study is a time-consuming process. Some classes of patients are particularly prone to drop out due to their age, disease condition or comorbidities. Many implantable devices require long-term follow-ups of five to eight years, and this, in turn, necessitates well-defined budgeting, planning and information storage strategies
How to Get It Right
Manufacturers should ensure that their PMCF studies are conducted in compliance with the international clinical investigations standard, ISO 14155. PMCF study designs should address the study’s objective, while the study hypothesis and endpoints should be scientifically sound, thus allowing valid conclusions to be drawn. At minimum, the PMCF study must define a rationale and justification of the chosen study, study population (corresponding to the CE-mark scope), objectives of study, related study endpoints and statistical considerations, inclusion/exclusion criteria, subject number and follow up period of patients, and justification for selection of sites and investigators.
#5 Lack of Communication with Notified Bodies
The Mistake
At present, the rate of certification of Notified Bodies (NBs) is not adequate for dealing with the new MDR demands, and the designation process for NBs is a lengthy one (see Exhibit 3). Certified NBs are unable to keep up with the numerous products backlogged in the recertification pipeline, and many manufacturers are waiting on their NB itself to recertify before resuming communications. A major PMCF mistake is not conferring with an NB about the adequacy of one’s study design to ensure MDR compliance.
The Consequences
NBs are liable to assess a manufacturer’s PMCF plans and their proposed execution, and thus require a thorough overview of all planned PMCF activities. Failing to establish a strong communication channel with the NB early on bears the risk of ending up with an inadequate study design which was not flagged on time. This, in turn, could result in costly modifications and time losses.
How to Get It Right
For maximum efficiency, manufacturers should initiate the PMCF plan review by their NB early on. This allows NBs to challenge any elements that will not stand up to scrutiny, thereby eliminating delays in the review process. For devices of lower classes, NBs may accept studies of simpler designs, reducing the threshold for patient participation and the costs to complete the investigation. An experienced and knowledgeable NB will clearly communicate with manufacturers and provide reasons as to why a plan may fail to meet MDR requirements.
#6 Managing PMCF on Paper or Spreadsheets
The Mistake
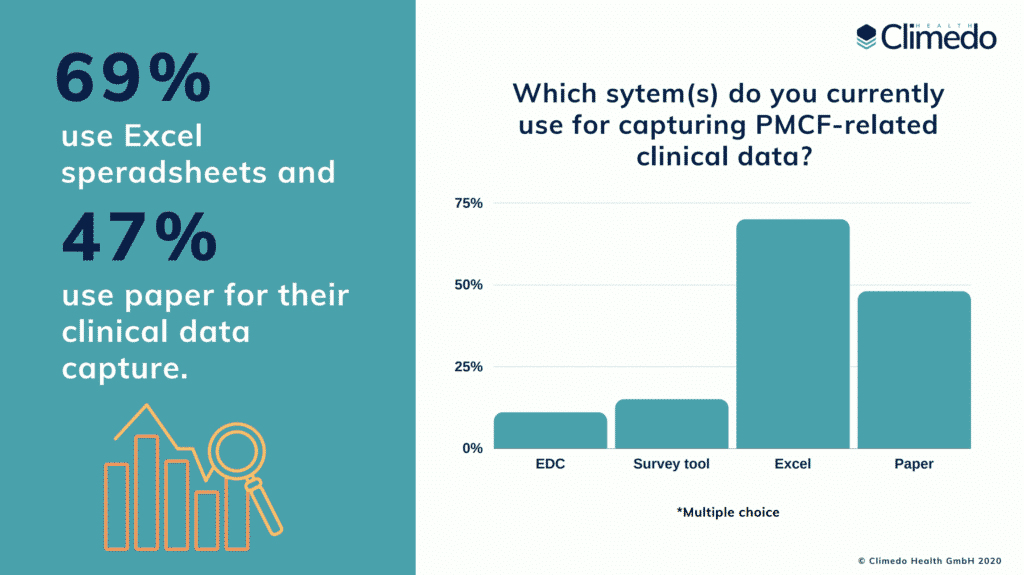
PMCF is a continuous process of collecting and updating clinical data in the post-market phase and requires an organized and scheduled system of planned annual activities. Many manufacturers do not yet have a logically organized and searchable PMCF system, and the various elements are spread out piecemeal over paper, email and spreadsheet processes. In fact, according to our EU MDR survey results on the true cost of the new regulation, 69% of MedTech companies still work with Excel and 47% work with paper. Just 11% use an EDC (electronic data capture) solution.
The Consequences
Since PMCF primarily measures residual risk after a device is on the market, any inadequacy at this level poses a serious risk. The time and resources required to complete PMS on paper are often significantly underestimated; as a result, projects can fail to meet deadlines, which in itself is a serious compliance issue. As a stop-gap, ineffective processes and expensive temporary fixes are implemented, putting an even greater strain on resources and personnel. Research suggests that the long-term costs of paper-based validation and clinical trials is actually up to 49-62% higher than for cloud-based solutions.
How to Get It Right
The PMCF plan documentation should be structured and presented in such a way that it facilitates its review and assessment by the NB. Organizations should abandon, paperbased processes, and legacy on-premise systems. Instead, they should collaborate with the right partners to help them automate their compliance processes. Not only will this help them reduce the administrative burden, it will support the more stringent traceability and vigilance required by the EU MDR.
Conclusion
Since Post-Market Surveillance must be a continuous and proactive process under MDR law, manufacturers will need to be more agile in their processes to keep their compliance up to date.
Manufacturers that set up PMS and PMCF plans using spreadsheets or paper will struggle to sustain efficiency and cost effectiveness in the post-EU MDR era. In the event of unannounced audits – which will become more regular under MDR law – paper-based firms will find it challenging to retrieve certain data and/or files on time.
Manual processes also require more resources to maintain, are more prone to human error and often require additional verification steps. Many time-consuming paper processes can be fully automated with an EDC system. Integrated surveys and ePRO / eDiaries with scheduled reminders allow both doctors and patients to report comfortably via email or SMS, thus improving response rates and reducing attrition. Using an EDC for validation and PMS is also a much safer solution, providing it is implemented using the highest level of data security.
Learn more about clinical data capture with Climedo in a free trial!

Dragan | Co-Founder & CTO
Climedo
Digital health entrepreneur. Passionate about clean UX and travelling to exotic countries. Creates products with love at Climedo Health.
You may also like these articles



