Unruhe wegen Freitag dem Dreizehnten? Vermeiden Sie diese 6 PMCF-Fehler!

DATUM
13. November 2020
AUTOR
Dragan | Co-Founder & CTO
Freitag der Dreizehnte muss kein Unglückstag sein. Falls Sie sich Sorgen machen, dass heute etwas mit Ihren klinischen Studien oder Umfragen schief laufen könnte, haben wir hier sechs typische PMCF-Fehler für Sie auf Lager, die Sie im Zuge der EU-MDR meiden sollten.
Vorwort
Nach der neuen europäischen Medizinprodukteverordnung (EU-MDR) wird die Post-Market-Überwachung (PMS) entscheidend für die Identifizierung und Bewertung potenzieller Restrisiken von Medizin-produkten auf dem Markt sein. Diese Risiken müssen Medizinprodukte-hersteller mithilfe eines systematischen Post-Market Clinical Follow-up (klinische Nachbeobachtung / PMCF) Plans nachverfolgen und bewerten. PMCF ist Teil des Qualitätsmanagementsystems (QMS) von Herstellern.
PMCF ist für alle Klassen von Medizinprodukten und Implantaten erforderlich. Mit der MDR-Übergangsfrist, die im Mai 2021 endet, müssen Hersteller für ihre Produkte nun proaktive PMCF-Studien durchführen. Dies gilt auch für Produkte, die bisher auf der Grundlage der Äquivalenz mit vergleichbaren Produkten auf dem Markt bewertet wurden. Um MDR-Konformität zu erreichen, müssen Restrisiken somit neu bewertet werden.
Zwar wird PMCF kurzfristig gesehen die firmeninternen Kosten erhöhen, kann aber auch langfristig als Chance zur Prozessanpassung für das gesamte Produktportfolio genutzt werden. Daher ist es wichtig, den Prozess richtig anzugehen. Ein proaktives PMCF-System trägt nicht nur zur Sammlung von Daten für die Einhaltung von Vorschriften bei, sondern kann auch zur Gewinnung umfassender Erkenntnisse und neuer Produktmarketing-Claims genutzt werden.
PMCF-Studien ähneln klinischen Untersuchungen und stehen oft vor vergleichbaren Herausforderungen.
6 PMCF-Fehler im Zuge der EU-MDR
- Laden Sie sich unser Playbook über typische PMCF-Fehler herunter
- Bereiten Sie sich auf mögliche Folgen vor
- Erfahren Sie, wie PMCF Ihnen gelingt

#1 Fehlinterpretation der Begriffe und Anforderungen
Der PMCF-Fehler
PMCF-Pläne umfassen ein breites Spektrum an Aktivitäten; dazu zählt für einige Unternehmen ggf. auch die komplett neue Aufsetzung einer PMCF-Studie. Hersteller verstehen oft die Unterschiede zwischen PMCF, PMCF-Plan und PMCF-Studien falsch oder verwenden sie beliebig.
Die Folgen
Eine Fehlinterpretation der Anforderungen kann zu unzureichenden bzw. inkorrekten Daten oder kostspieligen Verzögerungen bei der (Re- )Zertifizierung führen. PMCF ist ein breiter Begriff, der die Sammlung aller Arten von klinischen Daten wie etwa Vigilanz, Beschwerden, technische Informationen und öffentlich zugängliche Informationen umfasst. Die MEDDEV 2.7/1 rev. 4, die das primäre LeitfadenDokument für PMCF ist, legt jedoch einen großen Schwerpunkt auf die Durchführung von PMCF-Studien und die Datenerhebung.
Die Lösung
Woher weiß ein Hersteller, dass Marktdaten alleine evtl. nicht ausreichen und stattdessen eine vollständige PMCF-Studie erforderlich ist? MEDDEV 2.7/1 zeigt eine Reihe an Einschränkungen bei den Daten in der Pre-Market-Phase auf. Laut diesem Dokument zählen zu den Einschränkungen eine begrenzte Anzahl oder relative Homogenität der Probanden, eine geringe Vielfalt in der Studienpopulation oder ein Ungleichgewicht zwischen der Verwendung unter kontrollierten Variablen und der Verwendung unter allgemeinmedizinischen Bedingungen gibt. In diesen Fällen wäre PMCF also notwendig
#2 Das falsche Studiendesign
Der PMCF-Fehler
Wie bei jeder qualitativ hochwertigen klinischen Studie ist es auch bei PMCF-Studien wichtig, das korrekte Studiendesign zu wählen, um Daten richtig zu erfassen. Eine häufige Herausforderung bei Sponsorinitiierten Studien besteht darin, dass der Sponsor, in diesem Fall der Hersteller, womöglich nicht die vollständige Kontrolle über die Studiendurchführung hat, z.B. der korrekten Verwendung des Medizinprodukts innerhalb der Studienumgebung.
Die Folgen
Mit der bevorstehenden MDR-Frist werden zahlreiche PMCF-Studien durchgeführt, um die Konformitätsanforderungen zu erfüllen, insbesondere für Geräte der Klasse IIb oder höher. Daten aus diesen Studien müssen unbedingt als Konformitätsnachweis verwendet werden können. Wenn nicht, laufen Hersteller Gefahr, Geldstrafen einzubüßen, ihre Produkte vom Markt nehmen zu müssen oder keine Rezertifizierung zu erhalten. Dies kann vor allem zutreffen, wenn Hersteller bisher nur Äquivalenzdaten für die Zertifizierung nach der Medical Devices Directive (MDD) verwendet haben.
Die Lösung
Hersteller sollten ihre PMCF-Studien in enger Übereinstimmung mit der ISO 14155, dem internationalen Standard für klinische Prüfungen, durchführen. Das PMCF-Studiendesign sollte klar auf das Ziel der Studie ausgerichtet sein. Zudem sollten die Studienhypothese und Endpunkte wissenschaftlich fundiert sein, um valide Schlussfolgerungen zu ermöglichen. PMCF-Studien müssen mindestens folgendes definieren: Eine Rechtfertigung des gewählten Designs, die Studienpopulation (entsprechend dem CE-Geltungsbereich), die Studienziele, die damit verbundenen Studienendpunkte und statistischen Überlegungen, die Ein-/Ausschlusskriterien, die Anzahl der Probanden, die Nachbeobachtungszeit sowie die Begründung für die Auswahl der Prüfzentren und Prüfer.
#3 Die falschen Probanden
Der PMCF-Fehler
PMCF-Studien sollten sich auf den wichtigsten Anwender beziehen. So sollte etwa eine PMCF-Studie eines Ballonkatheters auf Feedback von Chirurgen basieren, da diese oft die einzigen Anwender sind. Umgekehrt sind bei „Wearables“ Patienten die wohl am besten geeigneten Probanden. Die Auswahl der falschen Studienpopulation ist oft auf mangelnde Erfahrung bei PMCF-Studien zurückzuführen. Oft werden Probanden auch nach den falschen Kriterien ausgewählt, etwa danach, wie gut erreichbar sie sind und nicht nach ihrer Vertrautheit mit dem Produkt.
Die Folgen
Die Auswahl einer falschen oder irrelevanten Studienpopulation für eine PMCF-Studie kann zeitaufwendig und kostspielig sein. Solchen Studien führen oft zu unzureichenden oder qualitativ schlechten Daten. Auch ist die Abbruchquote oft höher, was die Kosteneffizienz der PMCF-Aktivitäten weiter verringert.
Die Lösung
Unternehmen sollten sicherstellen, dass ihre gewählte Studienpopulation für die Erhebung wertvoller PMCF-Daten geeignet ist. Die strategische Auswahl von PMCF-Studienteilnehmer kann die Herausforderungen fehlender Daten oder niedrigen Rücklaufquoten minimieren, was wiederum die Kosteneffizienz der PMCF-Aktivitäten verbessert. Der Einsatz eines erfahrenen Teams inklusive Statistikern kann sich hier als guter Ansatz erweisen.
#4 Fehleinschätzung des Zeitaufwands für Patientenrekrutierung
Der PMCF-Fehler
Hersteller wissen oft nicht, dass PMCF-Studien (ähnlich wie klinische Untersuchungen) unter Herausforderungen bei der Patientenrekrutierung leiden können, insbesondere bei höher klassifizierten Produkten und wenn es um den Zugang zu den entsprechenden Patientenpopulationen und Zeitplänen geht.
Die Folgen
Das Screening und die Rekrutierung von Patienten ist fast immer ein zeitintensiver Prozess. Einige Patientengruppen sind aufgrund ihres Alters, ihres Krankheitszustands oder ihrer Begleiterkrankung besonders anfällig für einen Studienabbruch. Viele implantierbare Geräte erfordern Nachuntersuchungen von fünf bis acht Jahren, was wiederum eine fundierte Planung für Budgetierung und Datenmanagement erfordert. Eine unzureichende Planung führt zu Verzögerungen oder fehlenden Daten, was die Rezertifizierung gefährdet.
Die Lösung
Um mögliche Probleme frühzeitig zu erkennen, erfordern PMCF-Studien angemessene Strategien für die Nachbeobachtung von Probanden. Beispielsweise können Hersteller ihre Einschreibungsverfahren diversifizieren, um kosteneffizienter zu werden. Die Möglichkeit, Probanden online zu rekrutieren und dort ihre Einwilligung zu erhalten, wird die Zeitspanne erheblich verkürzen. Dies wird auch die Prozesse für die ethische Zulassung rationalisieren.
#5 Fehlende Kommunikation mit Benannten Stellen
Der PMCF-Fehler
Derzeit ist die Zertifizierungsrate von Benannten Stellen (“Notified Bodies” / “NBs”) nicht ausreichend, um den MDR-Anforderungen gerecht zu werden. Auch ist das Benennungsverfahren für die NBs relativ langwierig (s. Abbildung 3). Zertifizierte Benannte Stellen sind nicht in der Lage, mit den zahlreichen Produkten, die in der RezertifizierungsPipeline liegen, Schritt zu halten. Viele Hersteller warten auf die Rezertifizierung ihrer Benannten Stelle selbst und kommunizieren währenddessen nicht mit ihr. Eine unzureichende Kommunikation mit Benannten Stellen zum Designs von PMCF-Studien kann angesichts der EU-MDR zu großen Schwierigkeiten führen.
Die Folgen
NBs sind in der Pflicht, die PMCF-Pläne und vorgeschlagene PMCFDurchführung von Herstellern zu bewerten. Hierfür benötigen sie einen grundlegenden Überblick aller geplanten PMCF-Aktivitäten. Wenn es nicht gelingt, frühzeitig einen starken Kommunikationskanal mit der Benannten Stelle zu etablieren, kann ein ungeeignetes Studiendesign entstehen. Dies wiederum kann zu kostspieligen nachträglichen Anpassungen und Zeitverlusten führen.
Die Lösung
Um ein Maximum an Effizienz und Nutzen zu erreichen, sollte die Überprüfung des PMCF-Plans durch die Benannte Stelle frühzeitig initiiert werden. Eine Rückmeldung zu den vorgeschlagenen Plänen ermöglicht es der Benannten Stelle, alle Elemente, die der Prüfung nicht standhalten, in Frage zu stellen. Dies beugt Verzögerungen während des finalen Prüfungsprozesses vor. Insbesondere für Produkte niedrigerer Klassen können Benannte Stellen einfachere Studiendesigns akzeptieren. Dies reduziert die Schwelle für die Probandenteilnahme und die damit verbundenen Kosten. Eine erfahrene Benannte Stelle pflegt eine klare Kommunikation zu Herstellern und zeigt auf, warum ein PMCF-Plan ggf. nicht die MDR-Anforderungen erfüllt
#6 Durchführung von PMCF mit Papier oder Excel-Tabellen
Der PMCF-Fehler
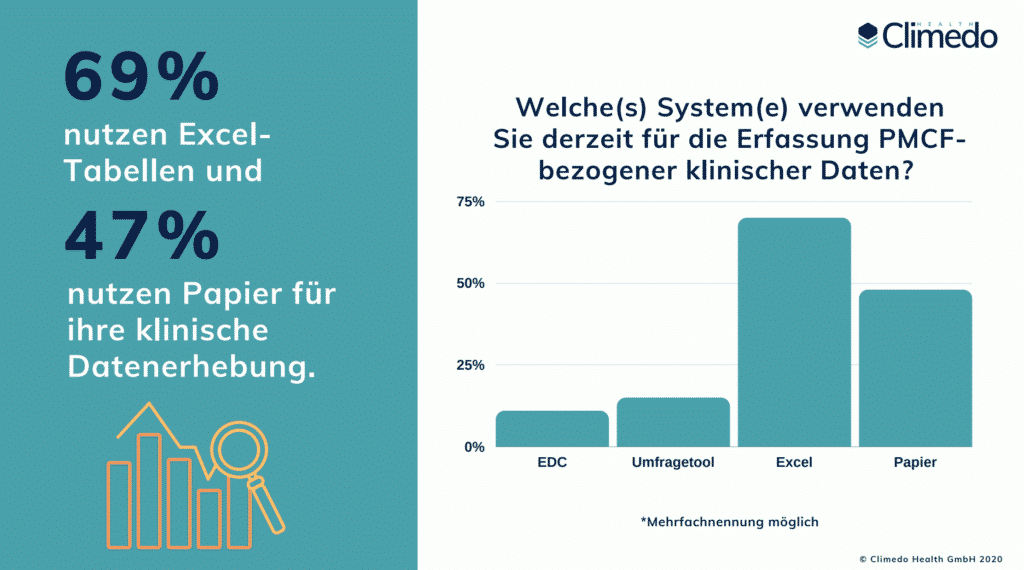
PMCF ist ein kontinuierlicher Prozess der klinischen Datenerhebung und -aktualisierung in der Post-Market-Phase und erfordert ein strukturiertes System von geplanten, jährlichen Aktivitäten. Viele Hersteller verfügen noch nicht über ein klar strukturiertes und durchsuchbares PMCF-System; die Elemente sind oft stückweise in Papierdokumenten, E-Mails und Excel-Tabellen verteilt. Unsere EU-MDR-Umfrageergebnisse zu den wahren Kosten der neuen Verordnung zeigten, dass 69% der befragten Hersteller noch Excel-Tabellen und 47% Papier für ihre klinische Datenerhebung nutzen. Erst 11% nutzen ein EDC (Electronic Data Capture) System.
Die Folgen
Da PMCF u.a. das Restrisiko eines Produkts nach dem Inverkehrbringen bewertet, stellt jeder Fehler auf dieser Ebene ein ernsthaftes Risiko dar. Der Zeit- und Ressourcenaufwand für die Durchführung von PMS-Aktivitäten auf Papier wird oft unterschätzt. Folglich werden Projektfristen nicht eingehalten, was wiederum ein Konformitätsproblem ist. Als Notlösung werden ineffektive Prozesse und kurzfristige Korrekturen eingesetzt, was Ressourcen und Personal noch mehr belastet. Studien zeigen, dass die langfristigen Kosten für papierbasierte Validierung und Studien tatsächlich bis zu 49-62% höher sind als bei cloud-basierten Lösungen.
Die Lösung
Die Dokumentation des PMCF-Plans sollte so strukturiert sein, dass sie die Überprüfung und Bewertung durch die Benannte Stelle erleichtert. Langfristig müssen Hersteller also papiergestützte Prozesse und veraltete Systeme aufgeben. Eine enge Zusammenarbeit mit digitalen Partnern wird es ihnen ermöglichen, ihre Konformitätsprozesse zu automatisieren. Dadurch verringern sie nicht nur ihren Verwaltungsaufwand, sondern kommen auch der von der EU-MDR geforderten strengere Rückverfolgbarkeit und Vigilanz nach.
Fazit
Da die Überwachung nach dem Inverkehrbringen laut EU-MDR ein kontinuierlicher und proaktiver Prozess sein muss, sollten Hersteller ihre Prozesse möglichst agil und digital gestalten. Damit wird es einfacher sein, sich an die neuen Vorschriften zu halten. Hersteller, die ihre PMS- und PMCF-Aktivitäten weiterhin auf Papier oder ExcelTabellen durchführen, werden sich schwer tun, effiziente Prozesse aufrechtzuerhalten. Im Falle unangekündigter Audits, welche unter dem MDRGesetz häufiger vorkommen werden, wird es für papierbasierte Firmen schwierig sein, bestimmte Daten und/oder Dateien rechtzeitig abzurufen. Manuelle Prozesse erfordern zudem mehr Ressourcen, sind fehleranfälliger und benötigen zusätzliche Überprüfungsschritte. Viele zeitaufwendige Papierprozesse können mit einem elektronischen Datenerfassungssystem (Electronic Data Capture / EDC) vollständig automatisiert werden. Integrierte Umfragen und ePRO / eDiaries mit geplanten Erinnerungen ermöglichen es Ärzten und Patienten, bequem per E-Mail oder SMS eine Umfrage auszufüllen. Dies verbessert die Antwortquote und reduziert die Abbrecherrate. Die Verwendung eines EDC für klinische Validierung und PMS/PMCF ist zudem eine wesentlich sicherere Lösung als Papier, sofern sie unter Verwendung des höchsten Maßes an Datensicherheit implementiert wird.
Erfahren Sie in einem kostenfreien Testzugang mehr über Ihre Möglichkeiten mit Climedo!

Dragan | Co-Founder & CTO
Climedo
Digital Health Unternehmer. Begeistert sich für sauberes UX und Reisen in exotische Länder. Produktentwicklung mit Herzblut.


