Wie wird die EU-MDR die Zukunft der MedTech-Branche verändern?

DATUM
15. Oktober 2019
AUTOR
Benjamin Sauer | VP Engineering
Ab dem 26. Mai 2020 müssen Medizinproduktehersteller innerhalb der EU die EU-MDR (EU 2017/745) einhalten. Ziel dieser neuen Verordnung ist es, die Patientensicherheit durch die Bewertung bestehender Geräte (z.B. durch PMS und PMCF) zu verbessern und die Transparenz über den gesamten Lebenszyklus eines Geräts zu gewährleisten. Bis zur Umsetzungsfrist der neuen Anforderungen bleiben nur wenige Monate, doch viele Hersteller sind noch ratlos darüber, wie genau sie sich vorbereiten sollten. In einer Umfrage von KPMG und RAPS gaben 66% der Befragten an, noch nicht mit der Planung für die langfristigen Auswirkungen der MDR begonnen zu haben. Nur 27% glaubten, dass sie bis Mai 2020 vollständig konform sein würden. Gründe hierfür sind unter anderem die relativ lange und komplexe Verordnung selbst, wenig Kommunikation von den zuständigen Behörden, ein Mangel an Benannten Stellen und die EUDAMED-Verzögerung, die jedoch die MDR-Frist selbst nicht betrifft.
Zeitenwende für die Medizintechnik
Trotz der Bürokratie und des zusätzlichen Zeit- und Kostenaufwands, die die neue Verordnung mit sich bringt, hat sie unserer Meinung nach das Potenzial, die MedTech-Industrie positiv zu verändern. Daher möchten wir nun ein wenig in die Zukunft blicken und fünf MDR-Aspekte analysieren, die die MedTech-Welt prägen werden und die Unternehmen sogar als wertvolle Wachstumshebel nutzen könnten
1. Beschleunigte Innovation
Die Post-Market-Überwachung (PMS) wird für die meisten Hersteller zum integralen Bestandteil ihres Qualitätsmanagementsystems (QMS). Damit müssen sie viel mehr Daten darüber erheben, ob ihre Geräte den versprochenen Zweck erfüllen. Zudem müssen sie leistungsschwache Geräte identifizieren und korrigieren oder beseitigen. Mit den neuen Datenquellen erhalten sie jedoch Zugang zu beispiellosen Erkenntnissen, mit denen sie statt reiner Konformität höhere Qualitätsstandards erreichen.
Darüber hinaus können Unternehmen von einer stärkeren Produktdifferenzierung und einer verbesserten Entwicklung und Vermarktung ihrer Geräte profitieren. Das Sammeln klinischer Bewertungsdaten und „real-world evidence“ zur Stützung von Produktversprechen kann zu neuen oder verbesserten Marketing Claims führen und so die Marktakzeptanz steigern.
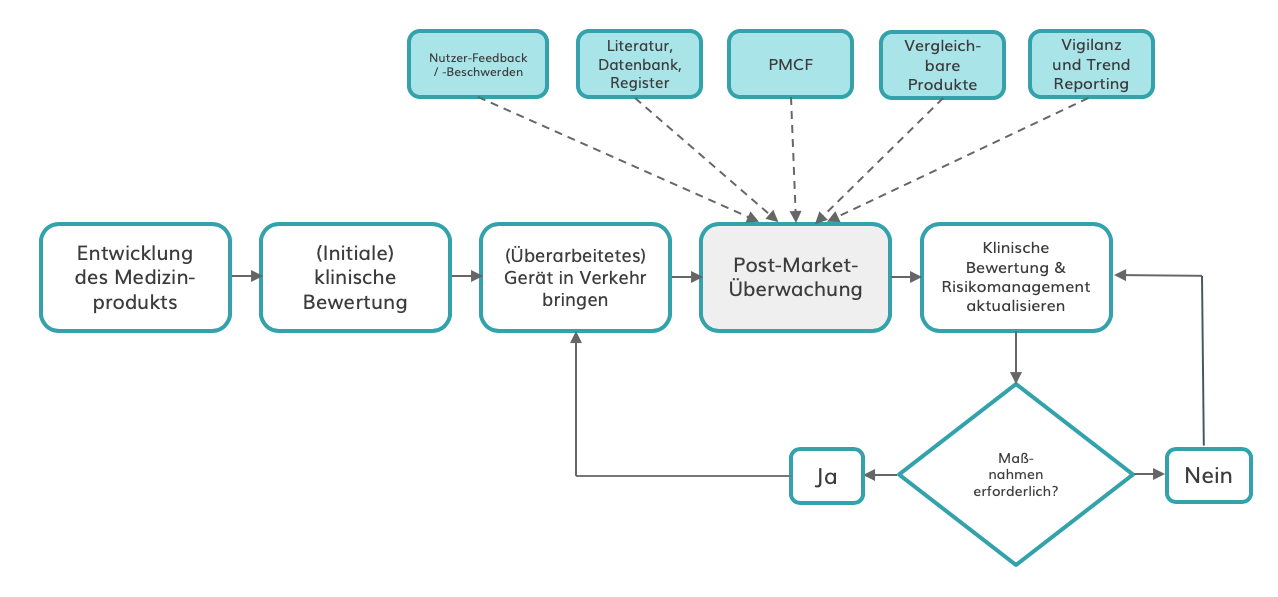
Verbesserter PMS-Prozess mit mehreren Informationsquellen
Quelle: Eigene Abbildung angelehnt am Johner Institut.
Experten zufolge könnten 30% der Medizinprodukte aufgrund der MDR vom Markt verschwinden. Obwohl dies beunruhigend klingt, wird es Unternehmen ermöglichen, ihre Ressourcen in ihre wertvollsten Geräte zu investieren und dadurch ihren Marktanteil zu erhöhen. Im Rahmen ihrer Portfoliobereinigung werden sie zudem erkennen, welche Produkte sich eventuell nicht mehr rentieren. Darüber hinaus wird die Unique Device Identification (UDI) helfen, Produktfälschungen zu identifizieren.
2. Mehr Sicherheit und Transparenz
Anders als die MDD (Medical Device Directive) erfordert die MDR im Artikel 10 (2) die Erstellung, Dokumentierung und Aufrechterhaltung eines Risikomanagementsystems (RMS) als Teil ihres QMS. Eine bessere Identifizierung, Analyse und Kommunikation bekannter und vorhersehbarer Gefahren wird langfristig eine verbesserte Patientensicherheit gewährleisten. Neue Sanktionen, wie etwa Geldbußen oder sogar Strafverfolgung, sollen sicherstellen, dass die Hersteller ihr RMS einhalten und das Wohlbefinden der Patienten in den Mittelpunkt stellen.
Die MDR kann zudem Haftungsrisiken verringern: Sollte die Sicherheit eines Medizinprodukts in Frage gestellt werden, müssen Hersteller schnell handeln. Dies trägt nicht nur zur Patientensicherheit bei, sondern reduziert auch die Haftung des Unternehmens, da die Reaktionszeit auf ein Minimum reduziert werden muss. Vorfälle müssen entweder sofort oder innerhalb eines bestimmten Zeitfensters (zwei bis 15 Tage) gemeldet werden. Die neuen Datenmengen, die bis dahin im Rahmen der PMS-Aktivitäten generiert wurden, werden bei Bedarf im Streitfall helfen, eine stärkere Argumentation aufzubauen
3. Patient Empowerment
Auf der Endverbraucherseite können Patienten und medizinisches Personal mit mehr Sicherheit, Rückverfolgbarkeit und Transparenz rechnen. Da Hersteller im Rahmen ihrer PMS proaktiv Nutzer-Feedback einsammeln und melden müssen, haben Patienten ein klares Mitspracherecht bei der Wirksamkeit von Produkten, wie sie angepasst werden, welche auf dem Markt bleiben und welche entfernt werden sollten. Dies wird zu einer Zukunft von wahrer „Patient Empowerment“ beitragen. Einige Beispiele dafür, was die MDR von Patienten herausfinden möchte, sind:
- Liefert das Gerät den versprochenen Nutzen?
- Wird ein Gerät evtl. zu häufig oder zu selten genutzt?
- Arbeiten Nutzer mit den Standardeinstellungen des Geräts oder passen sie diese immer selbst an?
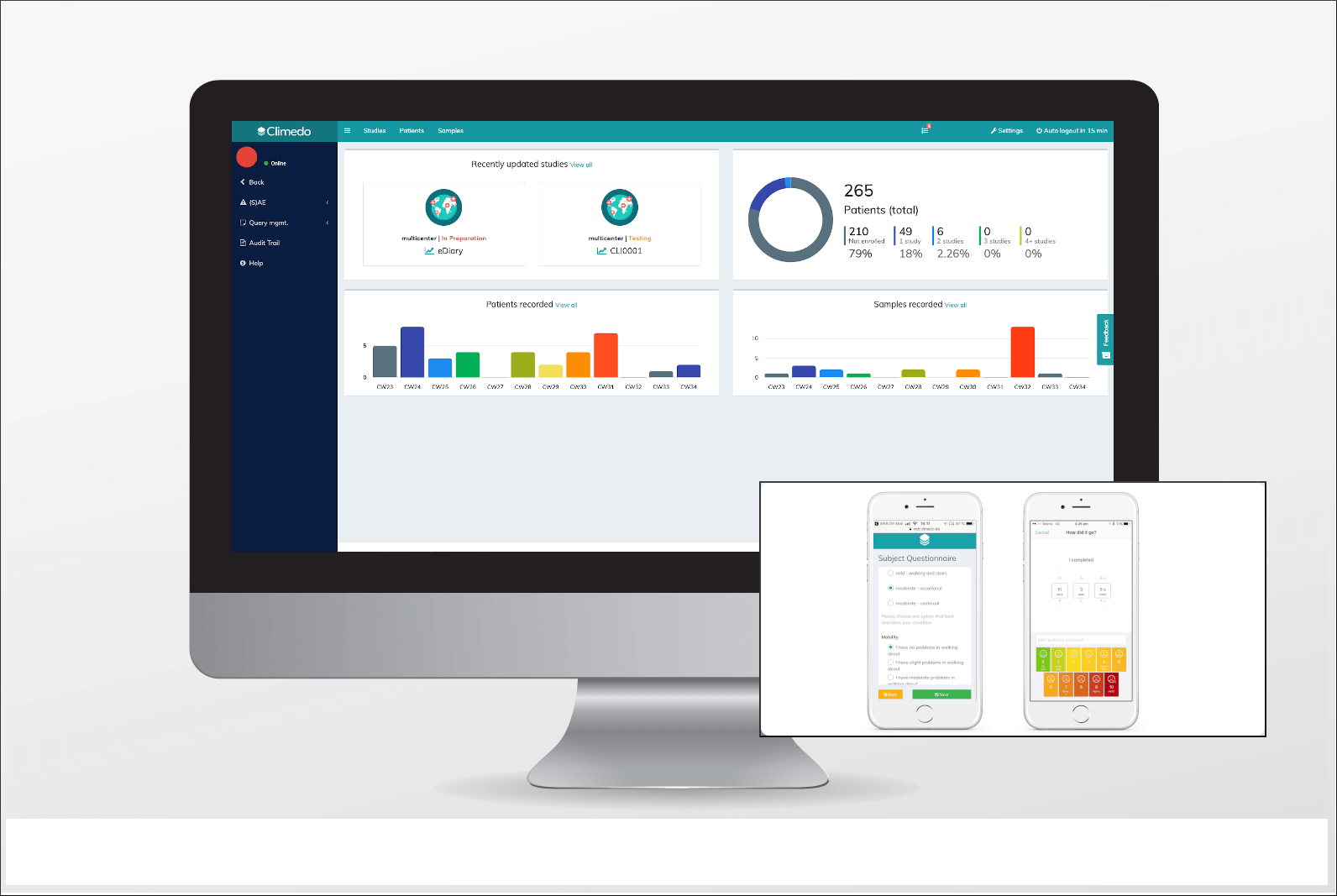
Beispiele von ePRO / eDiaries zur Erhebung von PMS-Daten durch den Patienten. Patienten können Umfragen per E-Mail oder SMS beantworten. Quelle: Climedo Software.
Mit der EUDAMED-Datenbank, welche im Mai 2022 öffentlich zugänglich wird, werden Patienten in der Lage sein, fundierte Entscheidungen über Geräte zu treffen, indem sie diese vor der Anwendung untersuchen. Patienten mit Implantaten erhalten zudem eine Registrierungskarte mit Zugang zu Informationen über den Hersteller und seine Sicherheitsbilanz.
4. Neue Rollen für Benannte Stellen
Medizinprodukte der Klassen IIa-III müssen von einer Benannten Stelle („Notified Body / NB“) überprüft werden. Als unabhängige Partei ist es ihre Aufgabe, sicherzustellen, dass die Geräte in jeder Phase, vom Entwurf über die Qualitätskontrolle bis hin zur Überwachung, der Verordnung entsprechen. So sind die benannten Stellen beispielsweise verpflichtet, die Regulierung durch unangekündigte Audits von Herstellungsprozessen durchzusetzen. Dadurch müssen Hersteller möglicherweise Verträge mit Lieferanten entsprechend anpassen.
Damit die benannten Stellen MDR-Zertifikate (wie z.B. die CE-Kennzeichnung) ausstellen können, müssen sie nach MDR neu zertifiziert werden. Dieser Benennungsprozess besteht aus vier Phasen und dauert ~18 Monate pro Stelle. Wir sollten daher mit einigen Verzögerungen im Überprüfungs- und Zertifizierungsprozess rechnen. Darüber hinaus ist es wahrscheinlich, dass nicht alle aktuellen benannten Stellen bestehen bleiben werden; die Zahl ist seit 2012 rückläufig – zum 20. November 2019 sind nur sieben davon MDR-zertifiziert. Die NANDO-Datenbank enthält eine aktuelle Liste.
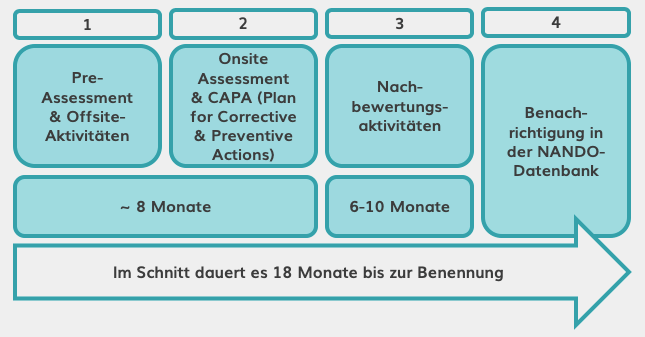
Benennungsprozess für benannte Stellen.
Quelle: Eigene Darstellung angelehnt an MedTech Europe.
Die strengere Überwachung und Kontrolle durch benannte Stellen wird dazu führen, dass Risiken durch unsichere Geräte drastisch reduziert werden. Hersteller sollten sich auf diese Änderungen vorbereiten und frühzeitig mit ihrer aktuellen Stelle sprechen, um einen reibungslosen Übergang zum MDR zu sichern.
5) Eine papierlose Welt
Laut Anhang II der MDR muss die technische Dokumentation von Herstellern einer „klare[n], organisierte[n], leicht durchsuchbare[n] und eindeutige[n] Form“ entsprechen. Viele betroffene Unternehmen arbeiten derzeit noch mit unstrukturierten, unflexiblen Datenerfassungs- und Freigabesystemen wie Papier, Excel oder E-Mail. Diese Systeme sind nicht nur kostenineffizient und unsicher, sondern werden in der Post-MDR-Ära nicht mithalten können. Daher ist mit einem Nutzungsanstieg von cloud-basierten Lösungen für die technische Dokumentation zu rechnen.
Immer mehr Hersteller werden somit auf eine ganzheitliche, digitale Lösung umsteigen, die alle Beteiligten vernetzt. Zu den Vorteilen zählen eine verbesserte Zusammenarbeit und Transparenz, die Abschaffung von Datensilos sowie effizientere Informationsmanagementprozesse. Zudem werden Unternehmen in der Lage sein, schneller auf schwere Vorfälle zu reagieren und mögliche Sicherheitsrisiken zu minimieren.
Um Daten vor Cyber-Risiken zu schützen, müssen jedoch gewisse Sicherheitsanforderungen erfüllt sein, wie etwa Daten-pseudonymisierung, geeignete Verschlüsselungsverfahren sowie konsistente Backup-Strategien.
Möchten Sie mehr erfahren?
Sollten Sie sich für eine persönliche, unverbindliche Demo unserer Software für automatisierte PMS interessieren, können Sie hier einen Termin vereinbaren.
Weitere EU-MDR-Inhalte zum Download
Infografik: 5 Schritte zur Vorbereitung auf die EU-MDR

Benjamin Sauer | VP Engineering
Climedo
VP Engineering mit der Mission, möglichst intelligente und benutzerfreundliche Software zu entwickeln, die den Alltag vereinfacht. Außerhalb der Arbeit Kampfsportler, Cineast, Rennradfahrer und Pflanzenliebhaber.
Das könnte Sie auch interessieren